{kind=link}
Новая химическая модель поможет точно рассчитать активность органических катализаторов
Российские ученые предложили новую модель для квантово-химических расчетов, которая позволит наиболее точно оценить активность органических веществ, ускоряющих химические реакции, в различных растворителях. Новый подход поможет быстро и без дополнительных затрат реактивов определять характеристики молекул, которые могут стать новыми эффективными катализаторами для синтеза лекарств и других биологически активных соединений. Результаты исследования, поддержанного грантом Российского научного фонда (РНФ), опубликованы в журнале Organic Chemistry Frontiers.
Современное химическое производство требует применения разнообразных высокоэффективных и в то же время экологичных катализаторов. Долгое время наиболее популярными были системы на основе металлов — очень активные, но загрязняющие окружающую среду, а также оставляющие примеси в продуктах реакции, что затрудняет синтез с их помощью химически чистых продуктов. Поэтому ученые ищут более безопасную альтернативу, на роль которой хорошо подходят органические молекулы, поскольку они по активности часто не уступают металлическим аналогам, и при этом не наносят вред окружающей среде.
Среди таких органических катализаторов самой высокой активностью обладают соединения, содержащие галогены — элементы 17 группы таблицы Менделеева (например хлор, бром и иод), — а также халькогены — элементы 16 группы, к которым относятся сера, селен и теллур. Эти вещества ускоряют химические реакции за счет того, что предоставляют «посадочные места» для чужих электронов — так называемые «сигма-дырки». Благодаря этому в присутствии катализатора электроны от соединений, вступающих в реакцию, стягиваются на эти «дырки», где остаются до того момента, пока нужное превращение не осуществится.
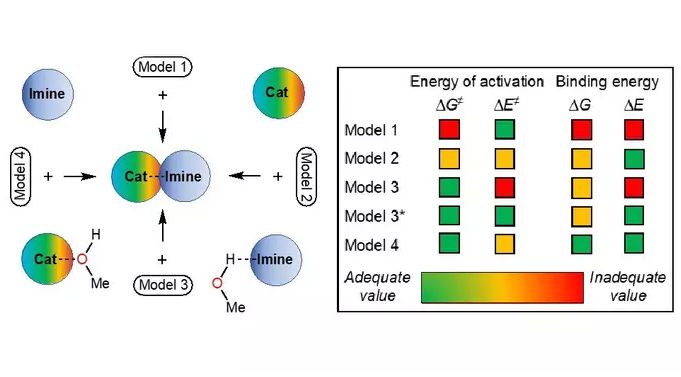
Ранее ученые из Санкт-Петербургского государственного университета (Санкт-Петербург) выяснили, что галоген- и халькогенсодержащие катализаторы имеют разную активность, которая зависит от строения молекулы. При этом оценить ее можно не только экспериментально, определяя скорость реакции, но и теоретически — с помощью расчета электростатического потенциала. Он показывает, насколько охотно вещество будет принимать электроны от других соединений. Такой подход до сих пор использовался только для сравнения относительной эффективности различных катализаторов, однако с его помощью было невозможно точно оценить абсолютную активность. В новой работе исследователи предложили модель для вычисления электростатического потенциала, которая позволяет решить эту задачу.
Авторы провели химическую реакцию, используемую в процессе синтеза некоторых лекарств и биологически активных соединений. Для ее ускорения использовали четыре иодсодержащих органических соединения, поскольку они, с одной стороны, высокоактивны, а с другой — позволяют получить очень чистые продукты, что крайне важно в медицинской химии.
Оказалось, что наиболее точно описать ход превращения позволила модель, в которой при расчете учитывалось взаимодействие исходного вещества не только с катализатором, но и с двумя молекулами растворителя, который служил для них своего рода окружающей средой. Эта поправка к расчету позволила химикам достоверно оценить активность каждого из четырех катализаторов. Правильность предложенной модели удалось подтвердить тем, что теоретические расчеты соответствовали наблюдаемым на практике скоростям реакции.